In this tutorial, I introduce a set of linux-based command line tools that perform the (i) Data Pre-Processing and (ii) Variant Discovery phases of the GATK Best Practices Somatic SNPs & Indels Workflow (pictured below), using O2's Linux-based HPC cluster, leveraging the SLURM job scheduler. Limited modifications will render these tools usable in conjunction with a variety of job schedulers, in addition to SLURM.
While the phrases "we performed Pre-Processing and Variant Discovery according to the GATK Best Practices" or we "we called variants using Varscan and Strelka", are incredibly straightforward, the fact of the matter is that at the time of this post's writing, executing a pipeline written by others in the genomics community requires careful integration of the pipeline with one's local or cloud HPC job-scheduling and file storage system. Members of the lab have collectively spent years writing scripts to execute the same standard genomics data processing pipelines. To save others precious time and effort, this series of tutorials serve to share well-documented and easy-to-use command line interfaces that execute various data processing pipelines.
Under the hood, the command-line tools presented use scripts that perfectly implement the Broad's GATK4 Best Practices, written in the Workflow Description Language (WDL), and execute them using the Cromwell Execution Engine. The scripts leverage a configuration file that encapsulates experimentation, troubleshooting and research that has yielded strong integration between the Cromwell Execution Engine and SLURM job scheduler to deliver consistently strong performance.
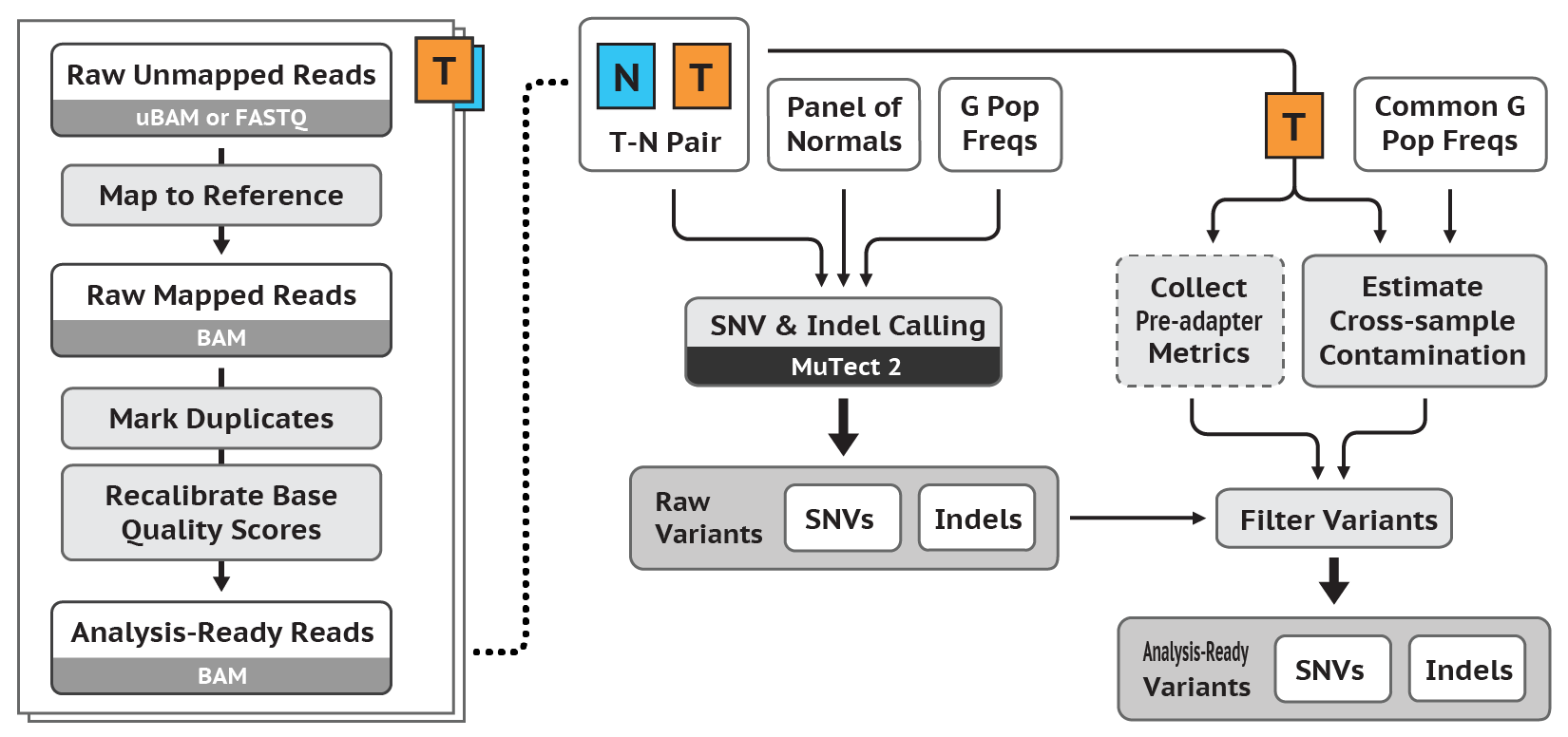
Please note that in contrast to the Germline SNPs & Indels Workflow Tutorial, this tutorial follows the workflow outlined in the previous version of GATK for (ii) Variant Discovery. It follows the tumor-normal workflow (it does not offer support for using a PON) and will be updated to the most recent version and include support for using a PON upon its subsequent revision.
For those completing the entire workflow, you will successively transform a number of samples of Raw Unmapped Reads to a set of Somatic SNPs & Indels (see above diagram).
The tutorial is purposefuly laconic - this is without a doubt a virtue and a consequence of the the ease-of-use of the command-line tools that follow, which encapsulate complexity and allow a user to minimize time spent running these tools and maximize their time reading and understanding the fundamental algorithmic methodology of the GATK Best Practices. You can read more about the methods and algorithms at work in of each of the phases of the GATK Best Practices Germline SNPs & Indels Workflow on the Broad Institute's website.
The scripts used in the tutorial can be accessed via the O2 file system in the following directory: /n/data1/hms/dbmi/park/SCRIPTS/GATK_Somatic_SNPs_Indels
Please note this is exactly the same workflow presented in Executing the GATK Best Practices Germline SNPS & Indels Workflow using an HPC cluster built on Linux and SLURM
Install Anaconda for Python 3 as per the instructions here.
Subsequently, create a new conda environment using the .yml file availible at /n/data1/hms/dbmi/park/alon/conda_envs/gatk.yml, using the following command:
conda env create -n gatk -f /n/data1/hms/dbmi/park/alon/conda_envs/gatk.yml
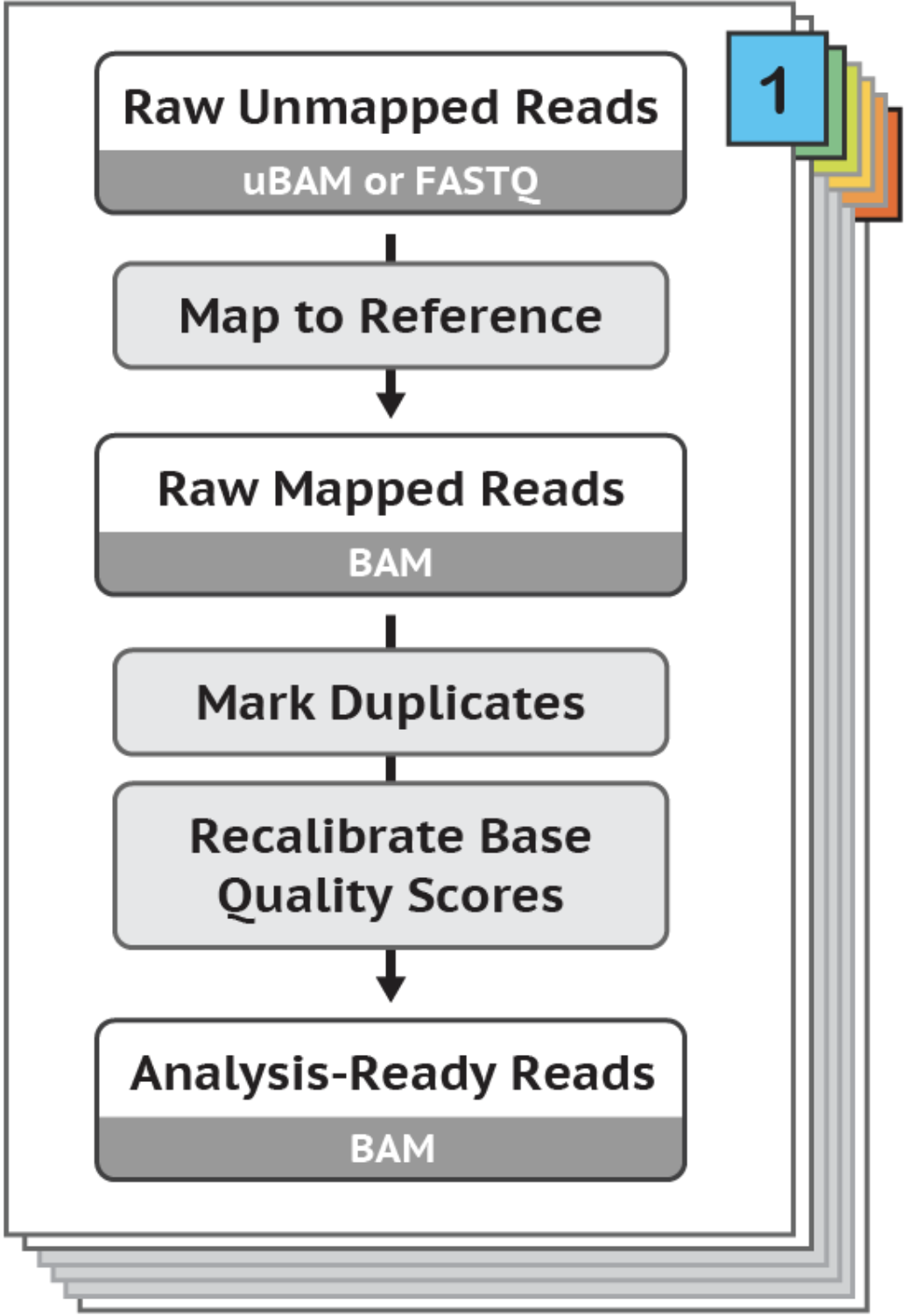
If your Raw Unmapped Reads are in FASTQ format, you must convert them to uBAM format, to yield Raw Unmapped uBAMs.
Use FastqToSam.py, which levarages Picard's FastqToSam to do this.
Example Command:
/path/to/FastqToSam.py --input_directory /my/input/directory --output_directory /my/output/directory
Use ValidateSamFile.py, which levarages Picard's ValidateSamFile to yield reciepts indicating whether or not a uBAM is Valid. Tool documentation included below:
Example Command:
/path/to/ValidateSamFile.py --input_directory /my/input/directory --output_directory /my/output/directory
Use CheckValidateSamFile.py to yield an assurance that all uBAMs passed ValidateSamFile. This tool simply scrapes the output of the previous command to achieve this goal.
Example Command:
/path/to/CheckValidateSamFile.py --input_directory /my/input/directory --output_directory /my/output/directory
Use PreProcessing.py to convert Raw Unmapped uBAMs to Analysis-Ready BAMs. This tool levarages a whole suite of GATK tools. Please see the default parameter of gatk4_data_processing_path, whose path can be determined by using the python PreProcessing.py --help command, to view the commands used.
Example Command:
/path/to/PreProcessing.py --input_directory /my/input/directory --output_directory /my/output/directory
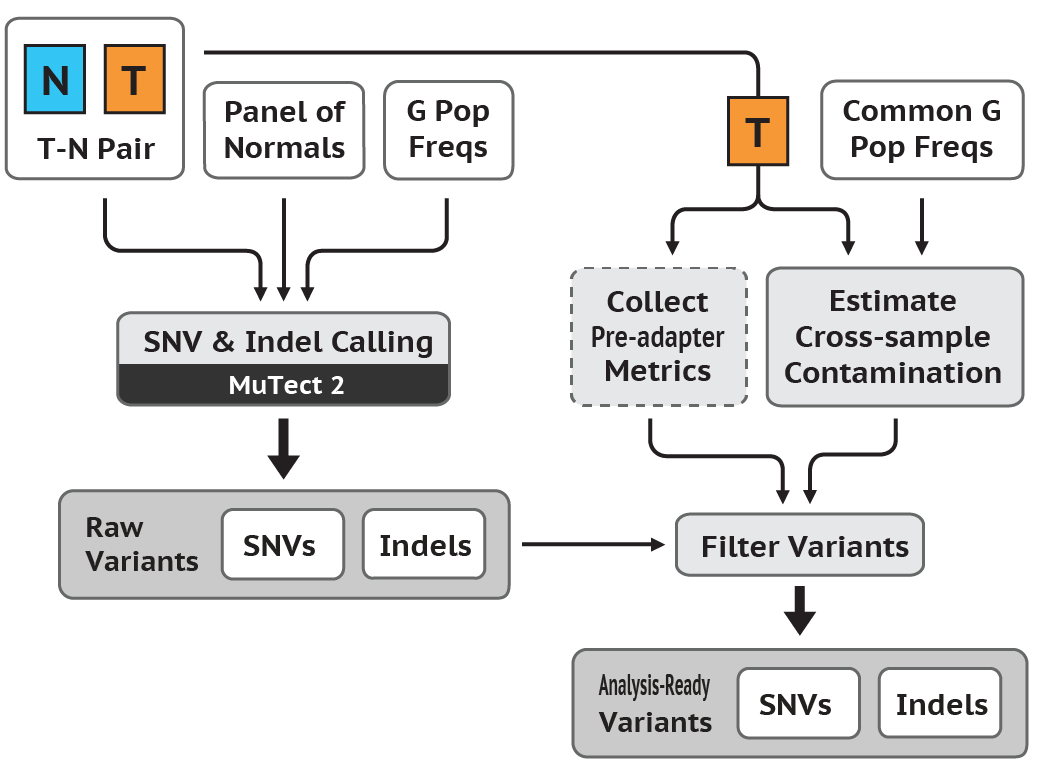
Use Mutect2.py, which levarages the Broad's Mutect2 to convert Analysis-Ready BAMs to VCFs.
Example Command:
/path/to/Mutect2.py -tumor /my/tumor/dir/tumor.bam -normal /my/normal/dir/normal.bam
Congratulations! You've executed the GATK Best Practices Somatic SNPs & Indels Workflow (i) Data Pre-Processing and (ii) Variant Discovery phases. Proceed to Callset Refinement!
$ python FastqToSam.py --help
usage: FastqToSam.py [-h] [-in_dir INPUT_DIRECTORY] [-out OUTPUT_DIRECTORY]
[-n NUM_CORES] [-t RUNTIME] [-p QUEUE]
[--mem_per_cpu MEM_PER_CPU] [--mail_type MAIL_TYPE]
[--mail_user MAIL_USER] [-picard PICARD_PATH]
[-library LIBRARY_NAME]
optional arguments:
-h, --help show this help message and exit
-in_dir INPUT_DIRECTORY, --input_directory INPUT_DIRECTORY
path to directory containing input files (default: ./)
-out OUTPUT_DIRECTORY, --output_directory OUTPUT_DIRECTORY
directory to which the "/.FastqToSam/" directory
containing outputs will be written to (default: ./)
-n NUM_CORES, --num_cores NUM_CORES
slurm job submission option (default: 1)
-t RUNTIME, --runtime RUNTIME
slurm job submission option (default: 2-0:00:00)
-p QUEUE, --queue QUEUE
slurm job submission option (default: park)
--mem_per_cpu MEM_PER_CPU
slurm job submission option (default: 10G)
--mail_type MAIL_TYPE
slurm job submission option (default: ALL)
--mail_user MAIL_USER
slurm job submission option (default:
email@hms.harvard.edu)
-picard PICARD_PATH, --picard_path PICARD_PATH
path to software (default:
/path/to/picard.jar)
-library LIBRARY_NAME, --library_name LIBRARY_NAME
name of the library the sample was prepared with
(default: lib_name)
$ python ValidateSamFile.py --help
usage: ValidateSamFile.py [-h] [-in_dir INPUT_DIRECTORY]
[-in_file INPUT_FILE_PATH] [-out OUTPUT_DIRECTORY]
[-n NUM_CORES] [-t RUNTIME] [-p QUEUE]
[--mem_per_cpu MEM_PER_CPU] [--mail_type MAIL_TYPE]
[--mail_user MAIL_USER] [-picard PICARD_PATH]
optional arguments:
-h, --help show this help message and exit
-in_dir INPUT_DIRECTORY, --input_directory INPUT_DIRECTORY
path to directory containing input files (default: ./)
-in_file INPUT_FILE_PATH, --input_file_path INPUT_FILE_PATH
path to input file (default: None)
-out OUTPUT_DIRECTORY, --output_directory OUTPUT_DIRECTORY
directory to which the "/.ValidateSamFile/" directory
containing outputs will be written (default: ./)
-n NUM_CORES, --num_cores NUM_CORES
slurm job submission option (default: 1)
-t RUNTIME, --runtime RUNTIME
slurm job submission option (default: 1-0:00:00)
-p QUEUE, --queue QUEUE
slurm job submission option (default: park)
--mem_per_cpu MEM_PER_CPU
slurm job submission option (default: 8G)
--mail_type MAIL_TYPE
slurm job submission option (default: ALL)
--mail_user MAIL_USER
slurm job submission option (default:
email@hms.harvard.edu)
-picard PICARD_PATH, --picard_path PICARD_PATH
path to software (default:
path/to/picard.jar)
$ python CheckValidateSamFile.py --help
usage: CheckValidateSamFile.py [-h] [-in_dir INPUT_DIRECTORY]
[-in_file INPUT_FILE_PATH]
[-out OUTPUT_DIRECTORY] [-n NUM_CORES]
[-t RUNTIME] [-p QUEUE]
[--mem_per_cpu MEM_PER_CPU]
[--mail_type MAIL_TYPE] [--mail_user MAIL_USER]
[-picard PICARD_PATH]
optional arguments:
-h, --help show this help message and exit
-in_dir INPUT_DIRECTORY, --input_directory INPUT_DIRECTORY
path to directory containing input files (default: ./)
-in_file INPUT_FILE_PATH, --input_file_path INPUT_FILE_PATH
path to input file (default: None)
-out OUTPUT_DIRECTORY, --output_directory OUTPUT_DIRECTORY
directory to which output will be written (default:
./)
-n NUM_CORES, --num_cores NUM_CORES
slurm job submission option (default: 1)
-t RUNTIME, --runtime RUNTIME
slurm job submission option (default: 0-01:00:00)
-p QUEUE, --queue QUEUE
slurm job submission option (default: priopark)
--mem_per_cpu MEM_PER_CPU
slurm job submission option (default: 8G)
--mail_type MAIL_TYPE
slurm job submission option (default: ALL)
--mail_user MAIL_USER
slurm job submission option (default:
email@hms.harvard.edu)
-picard PICARD_PATH, --picard_path PICARD_PATH
path to software (default:
path/to/picard.jar)
$ python PreProcessing.py --help
usage: PreProcessing.py [-h] [-in_dir INPUT_DIRECTORY]
[-in_file INPUT_FILE_PATH] [-out OUTPUT_DIRECTORY]
[-n NUM_CORES] [-t RUNTIME] [-p QUEUE]
[--mem_per_cpu MEM_PER_CPU] [--mail_type MAIL_TYPE]
[--mail_user MAIL_USER] [-overrides OVERRIDES_PATH]
[-cromwell CROMWELL_PATH]
[-gatk_wdl GATK4_DATA_PROCESSING_PATH]
[-input_json INPUT_JSON_PATH]
optional arguments:
-h, --help show this help message and exit
-in_dir INPUT_DIRECTORY, --input_directory INPUT_DIRECTORY
path to directory containing input files (default: ./)
-in_file INPUT_FILE_PATH, --input_file_path INPUT_FILE_PATH
path to input file (default: None)
-out OUTPUT_DIRECTORY, --output_directory OUTPUT_DIRECTORY
directory to which the "/.PreProcessing/" directory
containing outputs will be written to (default: ./)
-n NUM_CORES, --num_cores NUM_CORES
slurm job submission option (default: 1)
-t RUNTIME, --runtime RUNTIME
slurm job submission option (default: 30-0:00:00)
-p QUEUE, --queue QUEUE
slurm job submission option (default: park)
--mem_per_cpu MEM_PER_CPU
slurm job submission option (default: 10G)
--mail_type MAIL_TYPE
slurm job submission option (default: ALL)
--mail_user MAIL_USER
slurm job submission option (default:
email@hms.harvard.edu)
--orchestra_user ORCHESTRA_USER
slurm job monitoring option (default:
-overrides OVERRIDES_PATH, --overrides_path OVERRIDES_PATH
path to overrides.conf file (default: /path/to/overrides/overrides.jar)
-cromwell CROMWELL_PATH, --cromwell_path CROMWELL_PATH
path to cromwell.jar file (default:
path/to/cromwell-31.jar)
-gatk_wdl GATK4_DATA_PROCESSING_PATH, --gatk4_data_processing_path GATK4_DATA_PROCESSING_PATH
path to gatk4-data-processing file (default: /path/to/gatk4-data-processing.wdl)
-input_json INPUT_JSON_PATH, --input_json_path INPUT_JSON_PATH
path to gatk4-data-processing file (default: /path/to/input.json)
$ python Mutect2.py --help
usage: Mutect2.py [-h] [-tumor INPUT_TUMOR_PATH] [-normal INPUT_NORMAL_PATH]
[-out OUTPUT_DIRECTORY] [-n NUM_CORES] [-t RUNTIME]
[-p QUEUE] [--mem_per_cpu MEM_PER_CPU]
[--mail_type MAIL_TYPE] [--mail_user MAIL_USER]
[-gatk GATK_PATH] [-gatk4 GATK4_PATH]
[-reference REFERENCE_PATH] [-dbsnp DBSNP_PATH]
[-cosmic COSMIC_PATH] [-scatter SCATTER_SIZE]
optional arguments:
-h, --help show this help message and exit
-tumor INPUT_TUMOR_PATH, --input_tumor_path INPUT_TUMOR_PATH
path to input tumor file
-normal INPUT_NORMAL_PATH, --input_normal_path INPUT_NORMAL_PATH
path to normal file
-out OUTPUT_DIRECTORY, --output_directory OUTPUT_DIRECTORY
directory to which the output directory "/.Mutect2/"
will be written to
-n NUM_CORES, --num_cores NUM_CORES
slurm job submission option
-t RUNTIME, --runtime RUNTIME
slurm job submission option
-p QUEUE, --queue QUEUE
slurm job submission option
--mem_per_cpu MEM_PER_CPU
slurm job submission option
--mail_type MAIL_TYPE
slurm job submission option
--mail_user MAIL_USER
slurm job submission option
-gatk GATK_PATH, --gatk_path GATK_PATH
path to software
-gatk4 GATK4_PATH, --gatk4_path GATK4_PATH
path to software
-reference REFERENCE_PATH, --reference_path REFERENCE_PATH
path to reference_path file
-dbsnp DBSNP_PATH, --dbsnp_path DBSNP_PATH
path to dbsnp file
-cosmic COSMIC_PATH, --cosmic_path COSMIC_PATH
path to cosmic file
-scatter SCATTER_SIZE, --scatter_size SCATTER_SIZE